by
Brendon Nafziger, DOTmed News Associate Editor | April 28, 2010
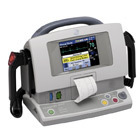
GE's Responder defibrillator (shown)
and other makes and models
have been added to the recall
The U.S. Food and Drug Administration expanded its recall of external defibrillators to include devices by GE Healthcare and Nihon Kohden, the agency said Tuesday, bringing the total number of affected defibrillators on the market to nearly 300,000.
The expanded recall is an update to one issued last November on some models of external defibrillators manufactured by Cardiac Science Corporation, and now includes GE's Responder and two Nihon Kohden models also made by Cardiac Science. Fourteen models are now affected, the FDA said.
"The FDA is issuing this notice so that users can take the proper steps necessary to assure they have access to safe and effective defibrillators," Dr. Jeffrey Shuren, director of the FDA's Center for Devices and Radiological Health, said in a statement.




Ad Statistics
Times Displayed: 3899
Times Visited: 9 Stay up to date with the latest training to fix, troubleshoot, and maintain your critical care devices. GE HealthCare offers multiple training formats to empower teams and expand knowledge, saving you time and money.
External defibrillators are machines found at airports, hospitals and homes that transmit electric jolts to revive patients stricken with sudden cardiac arrest. The recalled devices suffer from a "malfunction of the relay component and faulty resistors," an FDA spokeswoman told DOTmed News. Because of the manufacturing defects, the recalled devices might fail to deliver the life-saving shocks, according to the FDA.
The FDA said the device might also get interrupted while trying to monitor heart rhythms, and that it could be unable to recognize electrode placement.
Routine self-tests by the defibrillator's software might not detect the malfunction, and even a software fix issued by Cardiac in February does not detect all faulty electrical components, the FDA said.
Bothell, Wash.-based Cardiac Science first issued a recall on some Powerheart and Cardiovive models on November 13, 2009. While they issued a press release mentioning one of Nihon's models, called CardioLife and which is sold only in Japan, they did not explicitly link it to Nihon, an FDA spokeswoman said. The FDA's alert this week is the first to make the link and to name the GE device, she said.
The recall only affects devices manufactured between August 2003 and August 2009. Models affected are Powerheart 9300A, 9300C, 9300D, 9300E, 9300P, 9390A and 9390E; CardioVive 92531, 92532 and 92533; Nihon Kohden 9200G and 9231; and GE Responder 2019198 and 2023440.
The FDA recommends that most users switch to an alternative defibrillator if available or call Cardiac Science for repairs, but may continue to use the current device in an emergency. The agency advises high-risk facilities like hospitals to immediately get a new device or seek repairs.
Back to HCB News
















